

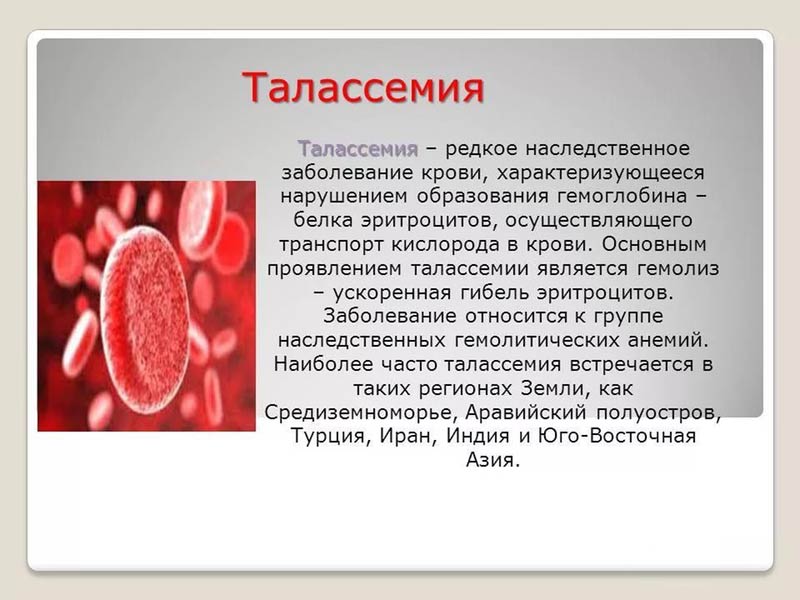
Талассемия
Талассемия (анемия Кули) — заболевание, наследуемое по рецессивному типу (двухаллельная система), в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина. В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров α-цепей и двух мономеров β-цепей. 3 % гемоглобина взрослых представлено гемоглобином А2, состоящем из двух альфа- и двух дельта-цепей. Существуют два гена HBA1 и HBA2, кодирующих мономер альфа, и один HBB-ген, кодирующий мономер бета. Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
В зависимости от того, синтез какого из мономеров нарушен, разделяют альфа-, бета- и дельта-талассемию. По тяжести клинических проявлений выделяют тяжёлую, среднюю и лёгкую формы заболевания.
Альфа-талассемия
Связана с мутациями в генах HBA1 и HBA2. Есть всего 4 локуса, кодирующего α-цепи. Наличие мутации в одном из локусов приводит к минимальным клиническим проявлениям. Нарушения в двух локусах выражаются лёгкой формой анемии. При мутациях в трёх локусах возникает значительное уменьшение продукции α-глобина. При этом избыточные цепи β-глобина образуют тетрамеры — гемоглобин Н. Эта форма носит также название гемоглобинопатии Н. Характер заболевания может варьироваться от лёгкой до тяжёлой картины гипохромной микроцитарной анемии. Присутствие мутаций во всех четырёх аллелях альфа-глобина не совместимо с жизнью. Ребёнок с такой патологией погибает внутриутробно или вскоре после рождения. Из пуповинной крови таких детей можно выделить гемоглобин Барта.
Бета-талассемия
Существует два варианта бета-талассемии — большая талассемия CD8(-AA) и малая талассемия (minor), из которых большая талассемия — наиболее тяжёлая форма заболевания. Возникает при наличии мутаций в обоих аллелях гена бета-глобина. В отсутствие или при резком уменьшении производства бета-цепей гемоглобин А вытесняется гемоглобином F, в норме вырабатывающимся у плода и сменяющимся на гемоглобин А после родов. Малая талассемия связана с наличием мутации в одном из аллелей гена бета-глобина. Как правило, протекает легко и не требует лечения.
Этиология
Талассемию вызывают точечные мутации или делеции в генах гемоглобина, ведущие к нарушению синтеза РНК, что приводит к уменьшению или полному прекращению синтеза одного из видов полипептидных цепей. Синтез цепей другого вида продолжается. Это приводит к образованию нестабильных полипептидных агрегатов из избыточных цепей, нарушающих нормальное функционирование эритроцитов и их разрушению. Повышенный гемолизэритроцитов вызывает анемию.
Эпидемиология
Альфа-талассемия распространена в Западной Африке и Южной Азии. Бета-талассемия часто встречается в странах Средиземноморья, Западной Азии и Северной Африки. Это регионы, где распространена малярия. Гетерозиготные носители мутаций в генах альфа- и бета цепей гемоглобина являются более устойчивыми к малярийному плазмодию. Имеются очаги талассемии в Азербайджане, в равнинных районах которого гетерозиготная бета-талассемия наблюдается у 7—10 % населения.
Клиническая картина
При талассемии характерны гипохромная анемия, анизоцитоз эритроцитов, наличие мишеневидных форм эритроцитов (пятно гемоглобина в центре клетки, напоминающее мишень). При этом содержание сывороточного железа нормальное или повышенное. Компенсаторная гиперплазия костного мозга, ведёт к нарушениям в строении лицевого черепа. Череп может стать квадратным, башенным; нос приобретает седловидную форму; нарушается прикус и расположение зубов. Отмечается желтушность кожи и слизистых оболочек. Селезёнка и печень увеличены. Больные подвержены инфекционным заболеваниям. Рано начавшаяся анемия обуславливает физическое и умственное недоразвитие ребёнка.


